Blog
Journal Club: Introducing Boltz-2, a State-of-the-Art Method for Predicting Protein–Ligand Complex Structures

At the first Journal Club held on April 2, 2026, we introduced the following paper.
“Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction”
Publication Venue
This paper was published on bioRxiv, a preprint repository for the biological sciences.
bioRxiv hosts research across a broad range of fields, including physiology, biochemistry, pharmacology, microbiology, genetics, and bioinformatics.
Overview of the Paper
This paper reports the development of Boltz-2, a new deep learning tool that can predict protein–ligand complex structures with high accuracy and speed, while also estimating binding affinity.
Understanding how a chemical binds to a protein is critically important in both drug discovery and toxicology. For that reason, methods that can accurately predict protein–ligand complex structures and binding affinities have attracted growing attention in recent years. The field of protein structure prediction has advanced rapidly since the emergence of AlphaFold2, which was later recognized by the 2024 Nobel Prize in Chemistry. Boltz-2 is one of the latest and most closely watched methods in this fast-moving area.
Boltz-2 stands out from existing approaches in three major ways:
(1) structure prediction accuracy,
(2) binding affinity prediction accuracy, and
(3) computational speed.
According to the benchmarks reported by the authors, Boltz-2 shows strong performance in predicting protein–ligand complex structures and delivers results that are competitive with AlphaFold3. The paper also reports favorable performance in terms of the physical plausibility of the predicted structures.
In addition, Boltz-2 demonstrates high performance in binding affinity prediction. One of its major advantages is its speed compared with conventional high-accuracy methods such as free energy perturbation (FEP). While FEP calculations may take hours to days, Boltz-2 has the potential to generate predictions in a much shorter time.
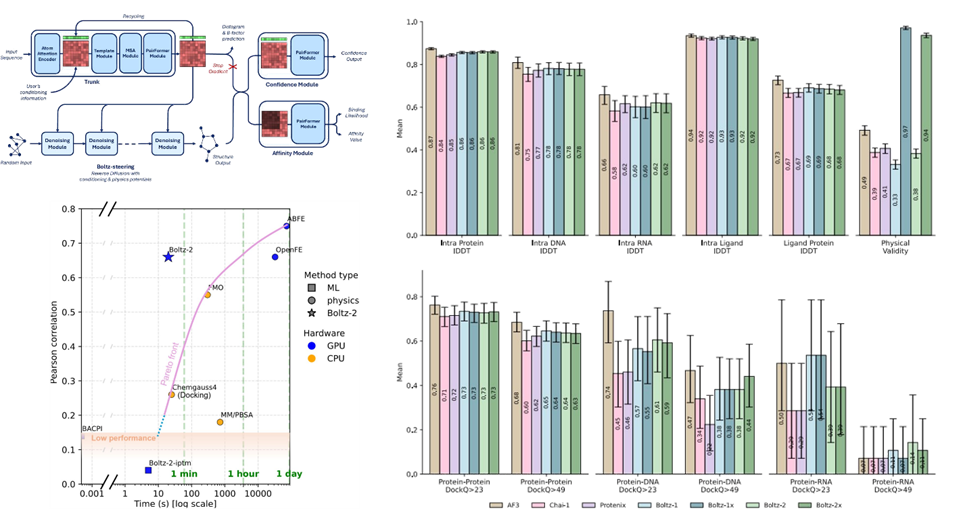
Figure. Architecture and benchmark results of Boltz-2. Adapted from Figures 1–3 in Passaro et al., “Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction,” bioRxiv (2025), DOI: 10.1101/2025.06.14.659707. Licensed under CC BY 4.0. Layout modified for this blog post.
Example Question from Lab Members
Q. What challenges remain before Boltz-2 can contribute to clinical applications?
A. Like simulation-based methods in general, Boltz-2 is more likely to be used at the screening stage rather than directly in clinical settings. In drug discovery and toxicity assessment, researchers first need to narrow down large numbers of candidate compounds. If protein–ligand complex structures and binding affinities can be predicted accurately and quickly at this stage, the experimental process can become more efficient and less costly, potentially improving the success rate of downstream development.
Why I Chose This Paper
My research focuses on developing methods to uncover the unknown toxic effects of chemicals by using structure-based simulations, including deep learning tools such as Boltz-2 and molecular docking. In understanding the mechanisms of chemical toxicity, both the protein–ligand complex structure and the binding affinity to target proteins are important factors.
Boltz-2 is particularly interesting because it integrates insights from structural biology into a deep learning framework to predict both structure and affinity with high accuracy and speed. I chose this paper because gaining a deeper understanding of the principles behind such a cutting-edge method will greatly contribute to the development of my own research.
In addition, Boltz-2 is already being used by some of the junior members of our lab. By discussing this paper in our Journal Club, I hoped to share the underlying concepts and principles of the method and thereby help raise the overall research standard of our laboratory.
References
Saro Passaro, Gabriele Corso, Regina Barzilay, et al.
Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction
bioRxiv 2025.06.14.659707
https://doi.org/10.1101/2025.06.14.659707
Jumper, J., Evans, R., Pritzel, A. et al.
Highly accurate protein structure prediction with AlphaFold
Nature 596, 583–589 (2021).
https://doi.org/10.1038/s41586-021-03819-2
Abramson, J., Adler, J., Dunger, J. et al.
Accurate structure prediction of biomolecular interactions with AlphaFold 3
Nature 630, 493–500 (2024).
https://doi.org/10.1038/s41586-024-07487-w
Latest Blog
Category
Archive
- 2026-4 (3)
- 2025-7 (6)
- 2025-6 (2)
- 2025-5 (2)
- 2025-4 (4)
- 2025-3 (4)
- 2025-2 (1)
- 2025-1 (2)
- 2024-12 (1)
- 2024-11 (3)
- 2024-10 (5)
- 2024-9 (1)
- 2024-8 (1)
- 2024-7 (4)
- 2024-6 (1)
- 2024-4 (1)
- 2024-2 (1)
- 2024-1 (1)
- 2023-11 (1)
- 2023-10 (1)
- 2023-6 (1)
- 2023-4 (1)
- 2023-3 (1)
- 2022-12 (1)
- 2022-11 (1)
- 2022-9 (1)
- 2022-6 (1)
- 2022-3 (3)
- 2022-1 (1)
- 2021-12 (3)
- 2021-11 (1)
- 2021-10 (1)
- 2021-9 (1)
- 2021-8 (1)
- 2021-7 (3)